Yoshikazu Furuta1,2, Hiroe Namba1, Tomoko F. Shibata3, Tomoaki Nishiyama4, Shuji Shigenobu3,5, Yutaka Suzuki1, Sumio Sugano1, Mitsuyasu Hasebe3,5, Ichizo Kobayashi1,2
(1 Medical Genome Sciences, Univ. Tokyo, Japan; 2 Institute of Medical Science, Univ. Tokyo; 3 National Institute for Basic Biology, Japan; 4 Kanazawa University, Japan; 5 Graduate University for Advanced Studies, Japan)
This poster describes a book project that aims to trace the important discoveries of 20th century tumor virology using oral history and archival sources. The thirteen proposed chapters cover the history from Peyton Rous’s discovery of Rous sarcoma virus to the creation of a vaccine for HPV and cervical cancer. Twenty seven major players in the history have been interviewed to date.
Narayana Annaluru1, Héloïse Muller1,2, Sivaprakash Ramalingam1, Joel Bader2, Jef D. Boeke2, Srinivasan Chandrasegaran1
1Department of Environmental Health Sciences, Johns Hopkins University School of Public Health, Baltimore, Maryland, USA
2High Throughput Biology Center, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
To enable the assembly of an entire synthetic yeast genome (Sc2.0 project), we have elaborated a workflow that allows us to design and synthesize pieces of DNA up to several kb starting from overlapping 60-mer oligonucleotides. The synthetic yeast genome was designed in such a way that the workflow is intrinsic to the design, which was done using a software suite called Biostudio. The wet-bench workflow consists of three distinct steps: In step one, 750 bp building blocks (BBs) are produced, which is carried out by JHU undergraduate students in the Build-A-Genome class, using PCR and standard cloning methods. In step two, Gibson isothermal assembly reaction was used for rapid and efficient fusion of several BBs into 3.0 kb DNA fragments (also known as minichunks) using a blend of three enzymes (exonuclease, polymerase and ligase). Alternatively, we used the yeast assembly protocol to fuse 3 to 5 adjoining BBs to generate several 3.0 kb minichunks. All consecutive minichunks were designed to have one BB overlap between adjacent partners. In step three, further assembly and replacement of the native DNA segments of chromosome III with synthetic fragments was carried out in vivo, using homologous recombination in yeast. Using this approach, we have successfully replaced the complete native chromosome III in S. cerevisiae, by its synthetic counterpart (synIII), using a LiOAc transformation protocol. Successive rounds of transformations using several overlapping minichunks with alternating genetic markers enabled us to create a synthetic chromosome, synIII. We propose that a similar strategy could be employed for the construction of a “designer” synthetic yeast genome for Sc2.0, in the future.
Desirazu N. Rao
Gajendradhar R. Dwivedi, Ritesh Kumar, Desirazu N. Rao
Department of Biochemistry, Indian Institute of Science, Bangalore – 560012, India
Helicobacter pylori is a highly genetically diverse bacterial species. It causes gastrointestinal diseases like atrophic gastritis, gastric adenocarcinoma, peptic ulcers and mucosa associated lymphomas. Amongst the three mechanisms of gene transfer in bacteria, natural transformation is a major cause of H. pylori high genetic diversity. Previously, a DNA binding protein, DprA (DNA Processing Protein A) was classified as recombination mediator protein owing to its role in protection of DNA and promoting RecA loading on it. In the present study, the molecular and biochemical role of DprA has been analysed in natural transformation pathway of Helicobacter pylori. It was observed that HpDprA (Helicobacter pylori DprA) binds and protects both ssDNA as well as dsDNA from various DNA nucleases. We noticed that not only HpDprA protected dsDNA from restriction enzymes but also stimulated the activity of methyltransferases on DNA. Greater protection from cognate restriction enzyme was observed when DNA was pre-methylated with methyltransferase in presence of HpDprA. Moreover, deletion of hpyAVIAM reduced the transformation efficiency. There was an increase in expression of restriction enzymes in hpyAVIAM deletion strains. These results combined together indicate that positive interaction of HpDprA with hpyAVIAM may result in epigenetic regulation of restriction enzymes. Thus, HpDprA inhibits the restriction enzymes by (a) occluding the restriction enzymes by substrate (b) modifying the substrate and thus rendering it resistant to restriction enzymes and (c) regulation of restriction enzyme expression in the cell. These results indicate that HpDprA could be one of the factors that modulate the restriction modification barrier during inter-strain natural transformation in H. pylori.
Analysis of secondary structure by CD spectroscopy showed that while DprA consists of a highly structured N-terminal domain the C-terminal domain on the other hand is random structured. Both DprA and NTD are dimers at higher concentrations and monomers at lower concentrations. Functional analysis of both these domains showed that NTD displays DNA binding property equivalent to full length protein. NTD stimulates activity of methyltransferase on DNA to a similar fold as that by DprA. Thus, NTD of DprA is responsible for its DNA binding activity and methyltransferase stimulation activity. However, further analysis is required to access in vivo role of CTD. Together, these data suggest that DprA could be a unique anti-restriction protein expressed by host bacteria to down-regulate its own R-M system to be able to support required genetic diversity during stress.
Marsha L. Richmond, Department of History, Wayne State University
While a full history of the discovery of restriction enzymes has yet to be written, men dominate the foundational narratives of this field. Women, however, also were central to the work that led to the discovery of restriction enzymes in the 1970s. Drawing on findings from the history of women in classical genetics, this work applies categories depicting women’s roles in genetics research, 1900-1940, to describe women’s participation in the history of restriction enzymes
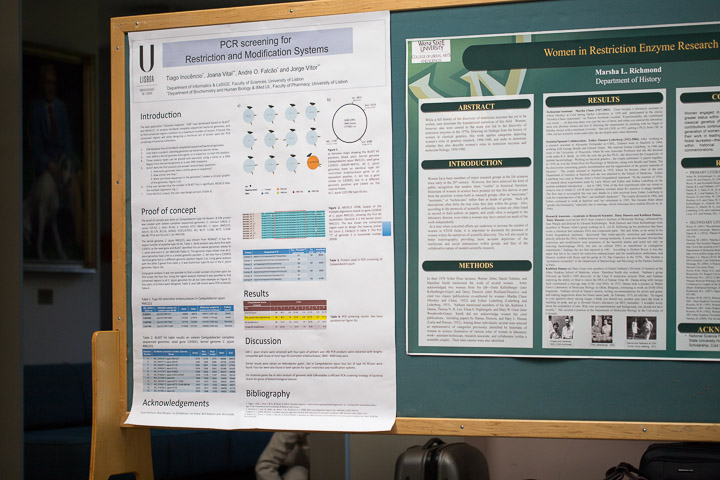
Tiago Inocencio*, Joana Vital**, Andre O. Falcao* and Jorge Vitor**
*Department of Informatics & LaSIGE, Faculty of Sciences, University of Lisbon, Portugal.
**Department of Biochemistry and Human Biology & iMed.UL, Faculty of Pharmacy, University of Lisbon, Portugal.
The microbiological/biochemical screening methodology for restriction and modification systems (RMS) has a major limitation: most microorganisms cannot be isolated and cultivated by current microbiological procedures. On the universe of cultivable microorganisms there are also some circumspections, the major one being the restraints on the use of pathogens for biomass production, for characterization or production of enzymes.
PCR is a remarkable technique, efficient and specific that amplifies DNA isolated directly from the environment, without the need to isolate and cultivate microorganisms. However it has a paramount handicap: can only be applied if the sequence of the gene of interest is known. This limitation can be overcome in some microbial genues: GenBank has more than 2500 complete sequenced microbial genomes and 21500 more in progress. We hypothesized that it would be possible to design a small number of primers pairs to PCR screening bacterial collections if RMS are flanked by conserved DNA sequences.
In order to test this hypothesis we developed a web application to find conserved genome regions common to a maximum number of strains: “Genome Inspector - GIN”. Based on BLAST and MUSCLE, GIN uses complete sequenced bacterial genomes deposited in Genbank or still unpublished (one file per strain, GenBank format). GIN employs a project-based approach in which users can select any given set of available genomes and perform a comprehensive bioinformatics analysis.
Here we present the results of PCR screening for Type IIG restriction endonuclease (REases) genes in a Campylobacter collection. The primers were designed after GIN analysis of sixteen genomes: C. concisus 13826, C. curvus 525.92, C. fetus 82-40, C. hominis ATCC BAA-381, C. jejuni (RM1221, 269.97, 81-176, 81116, IA3902, ICDCCJ07001, M1, NCTC 11168, NCTC 11168-BN148, PT14 and S3) and C. lari RM2100. Type IIG C. jejuni REases are present in four loci and eight highly conserved sequences were found flanking them.
Four pairs of primers were designed and 168 C. jejuni strains were screened. 340 PCR products were obtained with lengths compatible with those of most Type IIG restriction endonucleases, 3800 - 4000 base pairs. Once the extremities of the PCR products are sequenced, it is possible to design new primers to clone and express them. This strategy enables a safe and efficient screening of pathogenic bacterial strains for genes of biotechnological interest, and GIN plays a key role in this process.
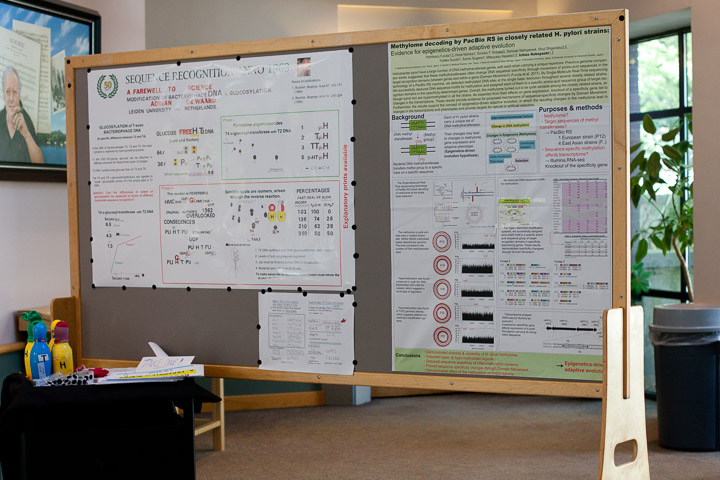
Adrian de Waard, Emeritus professor of the University of Leiden, the Netherlands.
The poster represents my earliest research as an independent investigator. Background: in the DNA of the bacteriophages T2,T4 and T6 the base cytosine is replaced by hydroxymethyl-cytocine (H). To this hydroxymethyl group a molecule of glucose can be attached, in different amounts for these three types of phages. T2 DNA contains less glucose than do T4 and T6. The T4 and T6 @ glucosyl transferases are capable to "super"glucosylate (some of ?) the empty sites in T2 DNA. Question: can the difference in specificity be explained in terms of a different nucleotide sequence recognition? I discovered that the @glucosyl transferase from T4 infected cells can differeniate between two nucleotide sequences: glcH-H and H-Pu. Other investigators claimed that this enzyme could glucosylate all of the sites that were devoid of glucose. I showed that they were misled by an artefact due to the reversibility of the enzyme, a phenomenon that they had established themselves. They had overlooked the consequences. These interchanges of radioactive label will be demonstrated with the help of models representing the various nucleotides.
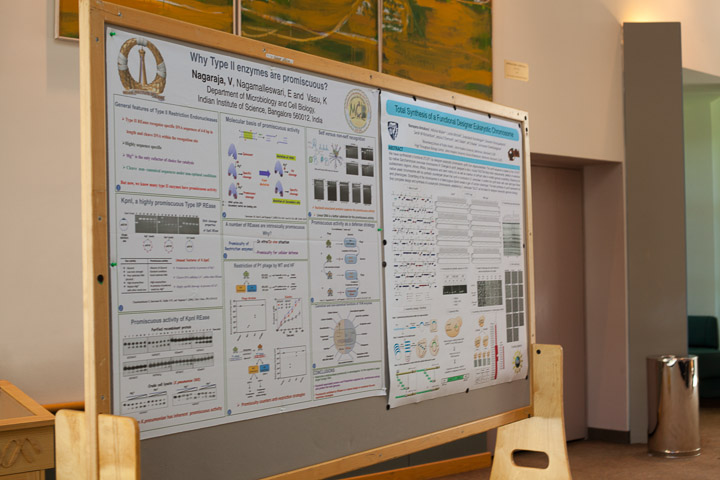
Nagaraja, V
Nagaraja, V, Nagamalleswari E and K. Vasu
Department of Microbiology and Cell Biology,
Indian Institute of Science, Bangalore 560012, India
From the time the restriction endonucleases were characterized, it has been generally accepted that they recognize and cleave DNA in a site specific fashion. The star activity detected in many of these enzymes was considered to be manifested either under unusual or non-optimal conditions such as ionic strength, glycerol, higher enzyme concentration etc. With the discovery of the highly promiscuous nature of Kpn1, an 'old' restriction enzyme, we have begun to ask more questions on evolution and intracellular roles of these enzymes. The enzyme exhibits promiscuity with respect to DNA binding and cleavage as well as co-factor utilization. The enzyme isolated from the Klebsiella pneumoniae OK8 strain exhibited promiscuous DNA cleavage similar to that of the recombinant, indicating that promiscuous activity is an inherent characteristic of the RE. In the work that followed, New England Biolabs have found a number of other enzymes to be promiscuous and converted them into high fidelity enzymes. Thus it is apparent that a number of RE are intrinsically promiscuous. Why?
There could be many reasons. One of them is a bacterial strategy to remain in the evolutionary 'arms race' with the invading genomes. The physiological basis for promiscuous behavior of Kpn1 was examined by classical phage titrations. Restriction of bacteriophages methylated at the canonical sequence indicated the functional relevance of promiscuity. Promiscuous DNA cleavage by KpnI thus appears to be a natural design of the organism, better equipped to degrade invading foreign DNA. The genome is protected by the topological state of the DNA, decoration by polyamines and binding by a variety of nucleoid associated proteins.
In addition, the intrinsic promiscuity in restriction enzymes may have other important functional base in diverse organisms. These points will be presented.
Alice Gutjahr, Shuang-yong Xu
New England Biolabs, Inc. Research Dept. 240 County Road, Ipswich, MA 01938, USA